Have a protein you want inhibited? New software can design a blocker
{kind=link}
Thanks in part to the large range of shapes they can adopt and the chemical environments those shapes create, proteins can perform an amazing number of functions. But there are many proteins we wish didn’t function quite so well, like the proteins on the surfaces of viruses that let them latch on to new cells or the damaged proteins that cause cancer cells to grow uncontrollably.
Ideally, we’d like to block the key sites on these proteins, limiting their ability to do harm. We’ve seen some progress in this area with the introduction of a number of small-molecule drugs, including one that appears effective against COVID-19. But that sort of drug development often results in chemicals that, for one reason or another, don’t make effective drugs.
Now, researchers announced they have created software that can design a separate protein that will stick to a target protein and potentially block its activity. The software was carefully designed to minimize the processing demands of a computationally complex process, and the whole thing benefits from our ability to do large-scale validation tests using molecular biology.
How do you calculate that?
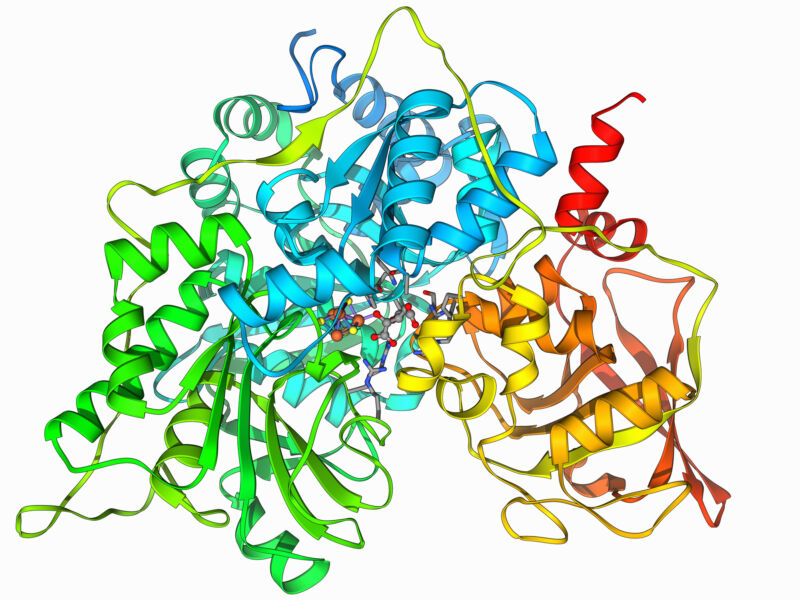
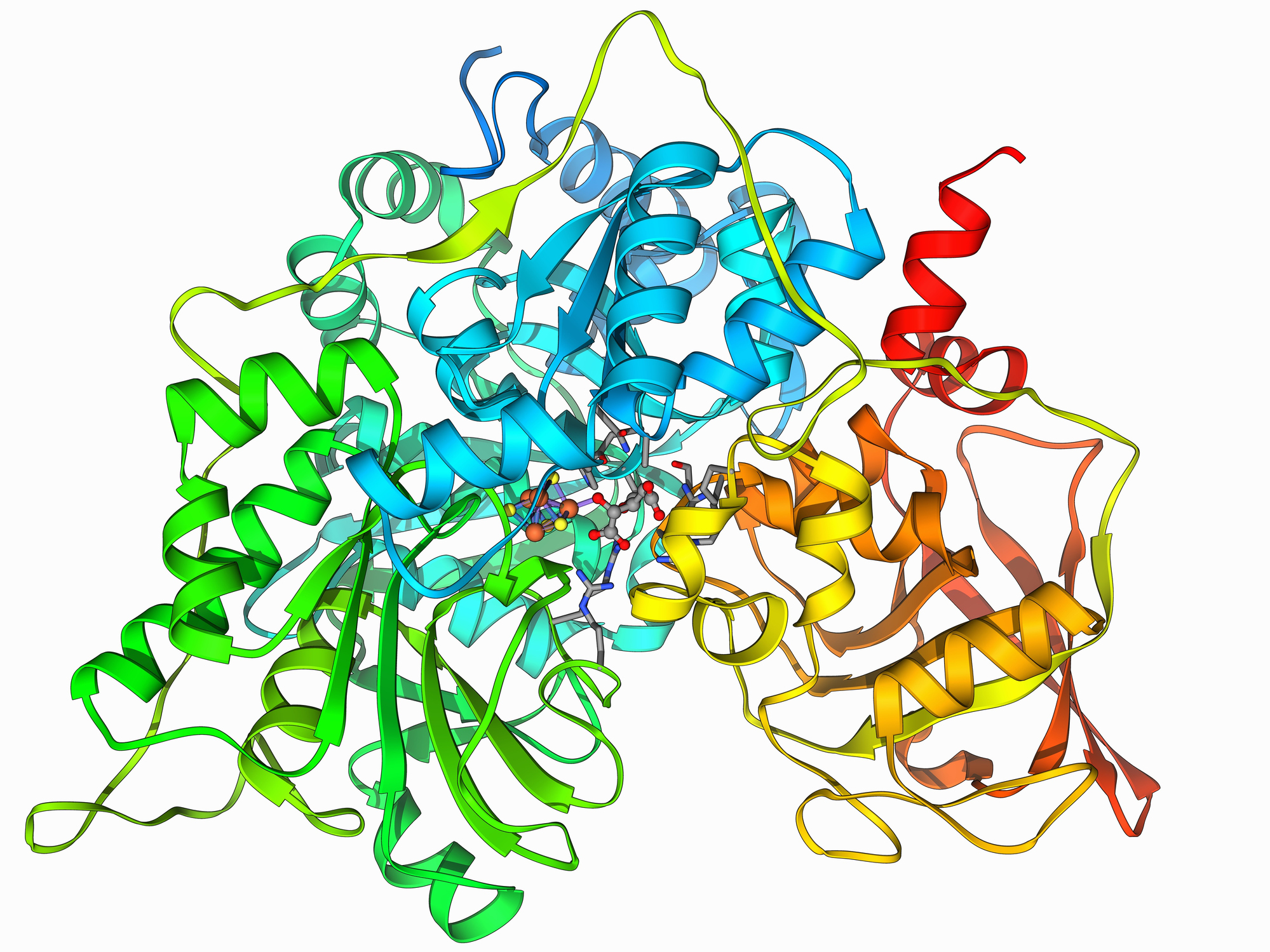
Proteins are a string of amino acids, with the cell using 20 different amino acids that have some distinct chemical properties. Interactions among these amino acids allow the proteins to adopt a three-dimensional configuration, which often brings key amino acids together in one location to form an “active site” that is critical for the protein’s function. Many drugs are designed to fit into this active site, which can keep a protein from working.
An alternative is to cover the site up with something big that sticks to it. And one of the easiest things to get to stick to a protein is another protein. This approach has some limitations when it comes to drug development. But the biggest problem is designing a protein that specifically sticks to another. Since each link in a protein’s chain can be occupied by one of 20 different amino acids, the number of possible proteins grows enormous after the chain is just a few links long. So exploring all possible interacting proteins is completely intractable as a computation.
The new work, which was done by a large US-based collaboration, focused on reducing the computational complexity. For the new process, the researchers decided to focus on producing lots of weak interactions rather than specifically designing a few strong ones. So the process started by taking each of the 20 individual amino acids and testing the strength of its interactions at billions of different sites on the surface of the target protein. The details of these interactions were then stored for later use.
Separately, the researchers built up a database of about 35,000 short proteins (about 50-65 amino acids long) that formed a stable core. These proteins were then placed next to the target protein. The algorithm would then determine where each amino acid was in close proximity to the target and then use the earlier result to pick which amino acid would have the strongest interactions at that location.
This process left a much smaller number of protein interactions that needed to be screened in detail. The researchers used a “packing” algorithm to see how the two proteins fit together and then estimated the strength of their binding.
https://arstechnica.com/?p=1843629